back (07.02.03/03.07.08*)
 DrTim homepage link
DrTim homepage link
Menopause - Hormones and Cancer (MHC)
Outline and Comments
by Timothy D. Bilash MD, MS, OBGYN
June 2003
based on the
Proceedings of the Second International Symposium
Portuguese Menopausal Society 1999
M. Neves-e-Castro and B.G. Wren (editors) 2002
(A remarkable conference about breast cancer mechanisms)
www.DrTimDelivers.com
>>
clic for
Highlights
/
Hormones & Cancer
.........
CHAPTER INDEX
- Aromatase and Estrogen BIOSYNTHESIS [CH 2 p9-13]
- Steroid RECEPTORS and Proliferation [CH 3 p15-22]
- The Role of ESTROGENS in Human Breast Cancer [CH 4 p23-36]
- New MECHANISMS for TIBILONE in Breast Cancer [CH 5 p37-47]
- Hormone Replacement, TUMOR BIOLOGY, and Prognosis [CH6 p49-53]
- Hormonal Therapy FOLLOWING BREAST CANCER [CH 7 p55-66]
- PHYTOESTROGENS and Breast Cancer [CH 8 p67-115]
- PROGESTERONE Therapy and Breast Cancer [CH 13 p117-121]
- PROGESTINS and Breast Cancer Risk: State of the Controversy [CH 14 p123-128]
- Hormones and Epithelial OVARIAN CANCER [CH 15 p129-140]
- NEUROSTEROIDS (Steroid Synthesis and Actions) [CH 1 p1-7]
- WHERE ARE WE NOW?(2002) [CH 16 p141-145]
- References
I. AROMATASE AND ESTROGEN BIOSYNTHESIS
[in Menopause - Hormones and Cancer Proceedings]
edited by M. Neves-e-Castro and B.G. Wren 2002
[CH2 p9-13]
Authors
J.H.H. Thijssen, J. van de Ven, P.C. de Jong
and M.A. Blankenstein [MHC CH 2]
Department of Endocrinology
University Medical Center Utrecht
WKZ Hp KE3.139.2
P.O. Box 85090
3508 AB Utrecht
The Netherlands
1999
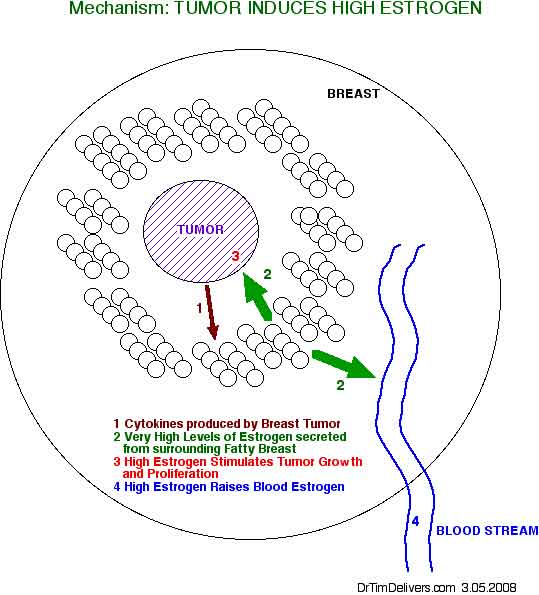
- there is no relationship between breast cancer and plasma levels of Estrogen!
- active uptake from the circulation plays no role in the accumulation of estradiol in tumors
- estrogens that stimulate tumor growth are converted locally in the fatty tissue surrounding the tumors from androgen precursors [MHC p11]
- postmenopausal, serum-to-tumor gradient exists for estrogen, particularly estradiol
- breast tumor concentrations of estrogen is same for pre- and postmenopausal women
- local estrogen contribution often exceeds 50%, especially in postmenopausal women
- most information we have is from model systems
- [ANOTE] There is evidence for a relationship between serum FREE Estradiol levels (FreeE2)
and Breast Cancer, but this model indicates the source is the fat tissue around the tumor,
not serum, and also levels of other blood components which affect Free Estrogen levels, such as Serum Binding Globulin (SBG)*.
- enzymes for conversion to estrogens are present in breast tumor and exist in the fatty tissue surrounding breast tumor
- includes 17HSD, aromatase, sulfotransferase, sulfatase
[see fig1 p10]
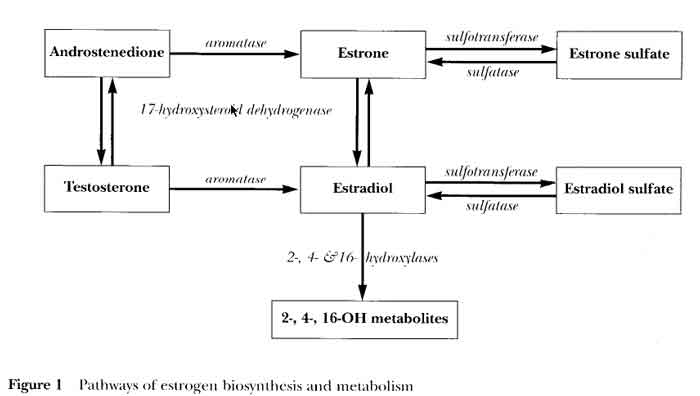
- 17HSD (17 Hydroxysteroid Dehydrogenase)
- Androstenedione (A) --> Testosterone (T)
- Estrone (E1).................--> Estradiol (E2)
- Aromatase
- Androstenedione --> Estrone (E1)
- Testosterone........--> Estradiol (E2)
- higher aromatase activity in fatty tissue suggests an interaction between tumor and adjacent adiopose tissue
- location has not been unequivocally determined
- using polyclonal antibody,
aromatase activity is found in stromal spindle cells and not in malignant epithelial cells.
- using monoclonal antibody and in situ hybridization to aromatase,
aromatase RNA expression was found mainly in malignant epithelial cells
- [ISLT] these distinct functions (aromatase activity and cell growth) are normally compartmentalized in different cell types, but abnormal cells exhibit multiple functions in the in same malignant cell
- aromatase inhibitors are first line treatment for metastatic breast disease
- decreased local and peripheral aromatase activity has been noted
- [ISLT] aromatase inhibitor would only inhibit enlargement of tumors, would not be curative
- Sulfatase
- Observations about estrogen and Breast Cancer
- Breast Ca incidence rises with age
- tumor hormone receptor positivity increases with age
- many tumors grow with estrogen, regress with no estrogen
- estradiol stimulates many cell growth factors
- Hypothesis: immune factors stimulate estrogen-dependent breast tumor growth
- interaction between malignant epithelial, stromal, adipocytes and macrophage cells
- evidence that tumor uses a different promotor for expression of aromatase than does adjacent adipose stromal tissue when producing estrogen
- Promotors II and I.3 are Tumor Promoters
- PGE2 is the main cytokine that stimulates cAMP and promotors II and I.3
- Promotor I.4 is a Normal Promoter for normal breast growth
- Class I Cytokines (Interleukin-6 / IL-6), Tumor Necrosis Factor Alpha (TNFa), in the presence of Glucocorticoids stimulates Promotor I.4
II. STEROID RECEPTORS AND
PROLIFERATION IN THE HUMAN BREAST
[in Menopause - Hormones and Cancer Proceedings]
edited by M. Neves-e-Castro and B.G. Wren 2002
[CH 3 p15-22]
Authors
R.B. Clarke and E. Anderson [MHC CH 3]
Clinical Research Department
Christie Hospital
Manchester M20 4BX
UK
1999
NORMAL BREAST, ESTROGEN & PROGESTERONE RECEPTORS
- Ovarian steroids are essential for the development, proliferation and differentiation of the breast
- Early menarch, late menopause increase the breast cancer risk
- early menopause protective from breast cancer
- pregnancy protective from breast cancer
- full lactational differentiation of breast epithelium in response to hormones in pregnancy
- breast changes with puberty in humans
- primary duct branching
- secondary duct branching
- terminal duct lobiloalveloar units (TDLU), intial
- Primary and Secondary ducts form a branching network
- double layer of epitehlial cells lining
- fibroblast layer
- extracellular matrix sourrounding
- TDLU (Terminal Duct Lobuloalveloar Unit)
- lined by luminal epithelium
- surrounded by basal and myoepithelial cells
- surrounded by basement membrane
- non-pregnant
- epithelial proliferation is maximal during the luteal phase (E and P effects from corpus luteum)
- during pregnancy
- development expands TDLU numbers
- after lactation, lobules involute back to non-pregnant appearance
- luminal cell is the major target cell for breast tumors
- Human vs Animal studies
- in mouse
- Estrogen induces growth of the ductal system during puberty (ductal elongation)
- Progesterone stimulates growth of the lobules in pregnancy (ductal side branching and alveolar development)
- in human
- lobules develop during puberty
- lobules mature and develop tumor resistance with pregnancy and lactation
- so mouse, human hormone studies may be different
- Normal human breast epithelium tissue proliferation rates
- human breast tissue transplanted into rats
- High Estrogen stimulates proliferation in normal breast tissue
- Progesterone has no obvious effects on proliferation in normal breast tissue
- Effects of Estradiol and Progesterone on Breast [see fig 2 p17]
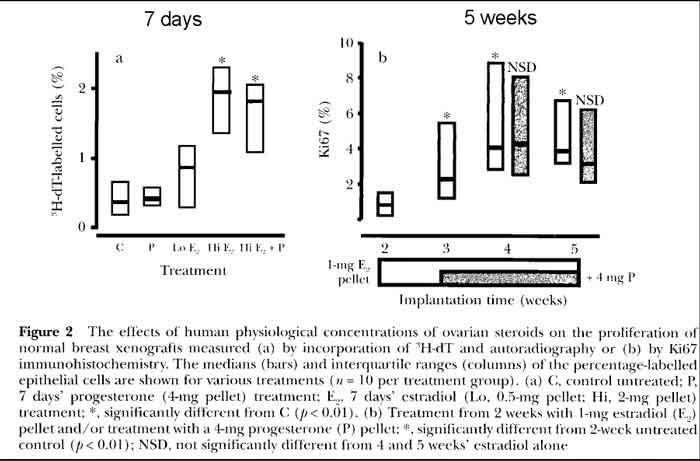
- P treatment of human breast cells
- Progesterone treatment alone shows very minimal proliferation
- low E treatment of human breast cells (follicular phase level)
- LOW Estrogen treatment shows minimal proliferation
- these estrogen levels are similar to HRT
- hi E treatment of human breast cells (luteal phase level)
- HI Estrogen treatment shows increased proliferation
- hi E+P treatment of human breast cells (luteal level E + P)
- HI Estrogen plus Progesterone shows same proliferation as HI estrogen only
- 5 week trial, short term
- In summary, a low dose of estrogen equivalent to follicular phase levels induced some proliferation, but
higher-dose luteal phase estrogen levels were necessary for a maximum induction of cell division; there were
no obvious effects of adding (natural) progesterone
- human breast tissue removed from women
- 5 years HRT of any type had no effect on proliferation rates [p18]
- > 5 years of combined HRT correlated with increased proliferation rates
- increased breast proliferation correlated with increased breast cancer risk
- Er, Pr receptors in normal breast
- E and P bind to receptors in the nucleus
- Er, Pr are transcription factors found within luminal epithelium cells
- not in myoepithelium or stroma cells
- 96% of cells that express one receptor express both
- Era, Erb subsets
- 10-15% of epithelial cells are Era, Pr positive
- distributed evenly in the interlobular and peripheral alveoli
- 2% of epithelial cells are proliferating, but do NOT contain E or P receptors
- proliferating (Ki67 antibody-positive) cells
- Pr and appear to be a separate population of cells
- proliferating cells do not contain Era receptors
- proliferating cells are often adjacent to or in close proximity to Era positive cells
- distinction between steroid receptor positive cells and proliferating cells is demonstrated in rats and mice
- 2 compartment model in normal breast (Clarke)
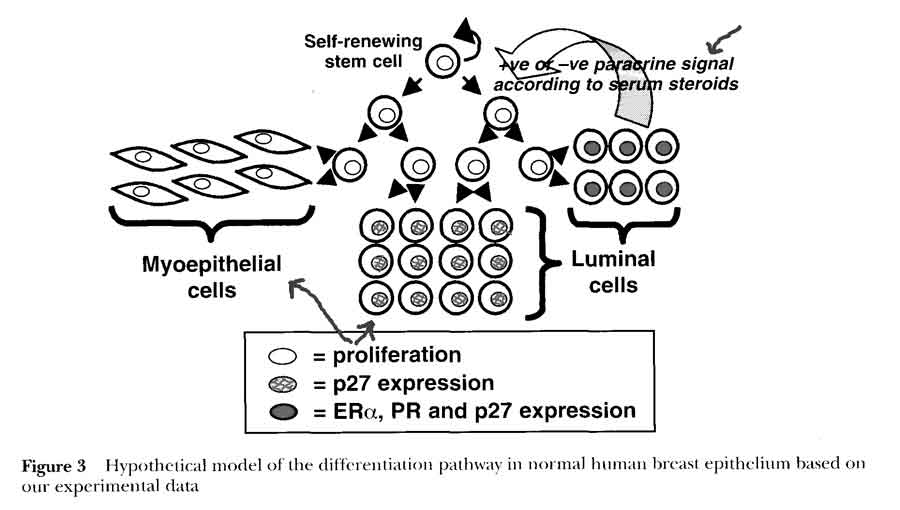
- HYPOTHESIS: steroid receptor-positive cells differentiate from a proliferative stem or transit cell population to act as steroid sensing cells and control cell proliferation via paracrine mechanisms
- 1st cells with receptors that sense and secrete hormones
- nearby 2nd cells without receptors proliferate due to steroid stimulation
- serum vs interstitial hormone levels (?comes from breast tissue)
- experimental evidence that receptors are not being down regulated during proliferation, but expressed in separate cells
- non-proliferating cells
- p27 antigen expressed once cells cease to proliferate
- expressed in terminally differentiated cells, including steroid receptor-positive cells
- Era receptor positive, act as estrogen sensors
- proliferating cells
- Ki67 antibody-positive
- very rarely express p27
- Era receptor negative (usually receptor negative)
- respond to positive or negative paracrine or juxtacrine factors secreted by Era cells in response to estradiol concentrations
- implies steroid secretion and proliferation are mutually exclusive activities in a cell [hormone secretion vs mitogen/growth factor effect]
- this separation of steroid receptor expression and proliferation in separate cells is disrupted at an early stage in tumorigenesis
- supports that estrogen is the major mitogen (not teratogen) in non-pregnant, premenopausal women
- progesterone may be more significant in post menopausal tumors
III. THE ROLE OF ESTROGENS IN HUMAN BREAST
CANCER: A MECHANISTIC VIEW
[in Menopause - Hormones and Cancer Proceedings]
edited by M. Neves-e-Castro and B.G. Wren 2002
[CH 4 p23-36]
Authors
J. Russo, MH Lareef, Q. Tahin, Y.F. Hu, C. Slater, I. H. Ruso [MHC CH 4]
Breast Cancer Reasearch Laboratory
Fow Chase CAncer Center
7701 Burholme Avenue
Philadelphia, PA 19111
USA
1999
Chromosome, Growth Studies
- Era (estrogen alpha) receptors
- reside in the nucleus in an inactive form associated with large inhibitory protein complexes
- all estrogens (endogenous and exogenous including DES) bind and activate the receptor pathways
- conformational change and dimerization takes place, causing binding to transcription factors (AP-1) which recruits co-activators (SRC-1)
- also forms ternary complex with a coactivator protein after interacting with regulatory sequences in promotor genes
- estrogen also has alternative non-receptor pathways
- overexpression of p21 induces Er and Er response promoters, in an estrogen-responsive manner without receptor
- Estradiol is converted to estrone, and both to catechol estrogens by cytochrome P450
- catechol estrogens induce an estrogen response
- not thru Er receptors (not inhibited by antiestrogen)
- estrogen may not need to activate its nuclear receptor to initiate or promote breast carcinogenesis
- estrogen and estrogen metabolites, P450 intermediates may exert direct genotoxic effects/ mutation
- still needs to be demonstrated in normal breast epithelium
- elevated synthesis or monomethylation lead to semiquinones and to quinones both of which are electrophiles that may be carcinogenic reactive intermediates in peroxidase activation
- Human breast model experiment (HBEC MCF-10F)
- MCF-10 is spontaneously immortalized cell line,
lacking Era, Erb receptors
- Erb receptors are induced in cells transformed by chemical carcinogens
- treated with Estradiol or DES over 2 weeks (4 applications)
- Estradiol and DES showed some similar effects as MCF-10F cells transformed by benzapyrene (BP) or oncogenes (causes growth)
- Analysis of specific genome alterations indicative of neoplastic transformation
- microsatellites
are polymorphic markers used to map the gene loci
- highly polymorphic, very common
- origin not well established, may result from polymerase errors
- used to mark allelic losses present in specific gene regions of transformed clones
- LOS- loss of heterozygosity
>50% reduction in one of the heterozygous alleles at known pertinent loci affected in ductal hyperplasia, carcinoma in situ, invasive carcinoma
- BP treated cells did not exhibit LOH at any of the loci tested (chromosome 3 or 11)
- only clones DES-5 in chromosome 3, and clones E2-1, E2-2 in chromosome 11, exhibited LOH
- most frequent allelic loss
found with breast cancer is in chromosome 17
, not yet found with estrogen treatments
- deletions in 3p gene regions
are not considered specific for breast cancer, but implicated as a genetic event triggered by DES
- estradiol and BP were NOT seen to cause LOH at chromosome 3 sites as DES did
- suggestion of suppressor genes in 3p regions
- 3p21.3 LOH found more frequently in metastatic than primary tumors
- 3p21.1-14.2 region deletions seen frequently with in situ carcinoma, benign tumors, familial breast cancers and is associated with dysregulated cell proliferation rather than tumor progression
- chromosome 11
contains LOH regions in breast, colon and other cancers, possible supressor gene dysfunction affecting early onset breast cancer and metatstatic processes
- significance of LOH in these regions and effects of estradiol needs to be clarified
- phenomic alterations indicative of neoplastic transformation
- doubling time
- doubling time only slightly decreased with Estradiol and DES treatment (78/73 vs 93)
- doubling time markedly decreased >50% with BP treatment (42 vs 93)
- [see tab 1 p26] markers of cell transformation
- colony counts
- DES clone = 151
- BP clone = 89
- E2 clone = 24
- control didnt form colonies = 0
- ductulogenesis (a measure of more normal ductal/non-solid component of tumor growth)
- decreased in estradiol treated cells, as well as DES
- [see fig 2 pg25] ducts vs solid growth
- induction of anchorage-independent growth
- increased in estradiol and DES treated cells
- so, there is still a confusing picture
- gene effects
: were rare with estradiol- and DES- treated cells. LOH was a rare event manifested in different chromosomes and in only a few clones, and not at all in BP treated cells
- growth effects
: phenotypic changes were induced by both estrogens (17bE2, DES) and chemical carcinogen (BP)
- estradiol increases proliferation (without branching)
- short-term treatment with 17betaEstradiol or DES induces anchorage-independent growth and colony formation in agar-methocel, and reduced duct formation in collagen gel, phenoytype expressions of neoplastic transformation which are also induced by BP under the same culture conditions.
- however, BP (tumorogenic) decreases proliferation, so proliferation does not necessarily indicate carcinogenesis
- estradiol effects are small compared to tumor inducing BP or DES
IV. NEW MECHANISMS OF ACTION FOR
TIBOLONE IN BREAST CANCER,
APOPTOSIS
[in Menopause - Hormones and Cancer Proceedings]
edited by M. Neves-e-Castro and B.G. Wren 2002
[CH 5 p37-47]
Authors
H.J. Kloosterboer [MHC CH 5]
N.V. Organon
Research and Development Laboratories
5340 BH Oss
The Netherlands
1999
- Clear evidence that steroid hormones have a role in breast cancer is lacking because of lack of studies
- thinking has been that estrogen + progestin may affect breast more adversely
- progestin potentiates estrogen effect at low dose
- Apoptosis*** (this is a key idea)
- programmed cell death
- part of natural cell life cycle
- new important mechanism in breast cancer
- cancer cells dont grow faster, actually grow slower in advanced stage
- cancer cells fail to undergo the programmed death (apoptosis)
that normal cells do
- Receptors
- receptor binding types
- Estrogen receptors
- Estrogen receptor Era (required for breast development)
- Estrogen receptor Erb (no effect on breast development)
- Progestin receptors
- Progesterone receptors PrA & PrB
- only one of the two required for breast development (redundancy)
- not required for normal alveoli in all breast epithelium ??? [p37]
- Androgen
- Glucocorticoid
- Mineralcorticoid
- normally proliferating breast cells do not contain receptors for either estradiol or progesterone, whereas proliferating breast cancers cells do.
- hi proliferation of normal breast epithelial cells seen during the luteal phase in humans
- Tibolone
- used in Europe for menopause complaints and osteoporosis
- less pain and tenderness
- lower mammogram density
- exhibits estrogenic and progestogenic properties, but has different effects than other HRT preparations
- combined effects of tibolone and metabolites are similar in human, monkey (and rat except for endometrium)
- prevents bone loss thru Er comparable to estrogens
- antiandrogen did not affect bone sparing of tibolone
- bone effect thru Er
- ???androgen effect is thru progesterone receptor or tibolone metabolites
- treats hot flushes and vaginal atrophy
- Er effects of metabolites
- [see table 1 p39] tibilone receptor activation
- tibolone parent steroid
- tibolone has short half-life
- rapidly metabolized via 3BHSD (3b-hydroxysteroid dehydrogenase)
- not evident in high concentrations because of rapid metabolism
- tibolone not metabolized via pregnenolone
- [see fig 1 p38] metabolism of Tibilone
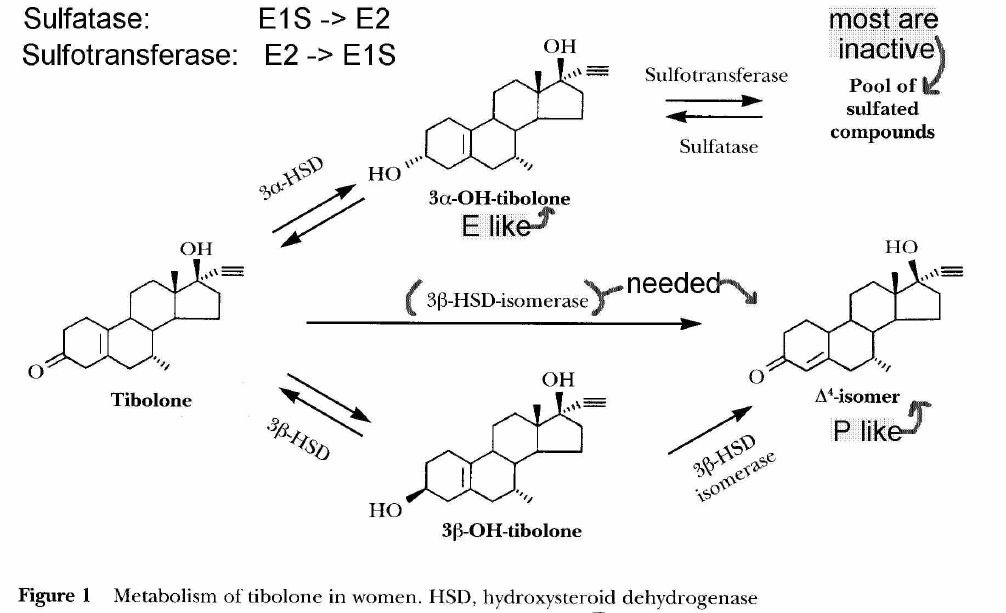
- doesnt activate Er receptor
- lacks aromatic A-ring with 3-OH group for Er binding
- binds Er only at high concentrations
- some binding to Progesterone (Pr) and Androgen (Ar) receptors
- lacks 3-keto-delta4 configuration for good binding to Pr and Ar
- tibolone activates Pr and Ar receptors
- 3a-,3b-(OH) tibolone metabolites
- Estrogenic
- 3a, 3b (OH) metabolites bind to Era and Erb receptors
- do not bind to Pr, androgen, glucocorticoid or mineralcorticoid receptors
- 3a-OH concentration 4x> 3b-OH
- Era 10x > Erb binding
- 7hr half-life
- mostly found in inactive sulfated form
- 3b sulfated form not usually found
- delta4-isomer tibolone metabolite
- Progestogenic activity with prolonged action
- long 1/2 life (7a-methyl group)
- compared to progesterone which is quickly inactivated by reduction of double bond in A ring
- Androgenic activity
- delta4 isomer binds, activates Pr, Ar receptors, but not Er, glucocorticoid or mineralcorticoid receptors
- in human endometrium, progestagenic activity of tibolone dominates
- due to local formation of the progestagenic delta4 isomer from 3b-(OH) tibolone metabolite
- completely inhibits proliferation of the endometrium in postmenopausal women and monkeys
- rat not good model for human endometrium
- tibolone preferentially metabolised to estrogenic 3OH-metabolites
- no local delta4 isomer in rats, estrogenic effect stimulates uterine growth
- but still anti-tumor in rat in spite of estrogenic properties
- progestogenic effects dominate as in OCP's
- longer half life (anti-estrogen effect)
- no breast stimulation seen with tibolone, in contrast to estrogen
- tibolone compared to antiestrogens, androgens which have different mechanisms
- tibolone/metabolites diminishes ligand levels for estrogen receptor (steroid levels)
- antiestrogens (tamoxifen) modify the estrogen receptor itself
- Androgens have an antiestrogenic effect on the breast
- mixed estrogen effect, thru RNA steroid effects in cytoplasm and not receptors
- but tibolone effect on breast not directly thru androgen receptors
- [ISLT] could still have an indirect androgen effect thru 17BHSD and delta4 isomer
- PEPI trial- medroxyprogesterone actetate and progestin
- women receiving conjugated estrogen plus progestin had an increase in mammographic density (20% medroxyprogesterone acetate, 16% micronized progesterone), indicating more stroma or ductal and glandular tissue
- Almost all increases in mammographic density occurred within the first year
- both cyclic and continuous E/P dosing
- 0% (CI= 0.0% to 4.6%) in the placebo group
- 3.5% (CI= 1.0% to 12.0%) in the CEE group
- 23.5% (CI= 11.9% to 35.1%) in the CEE plus cyclic MPA group
- 19.4% (CI= 9.9% to 28.9%) in the CEE plus daily MPA group
- 16.4% (CI= 6.6% to 26.2%) in the CEE plus cyclic MP
- DMBA model on human breast
- tibolone reduced tumor growth similar to tamoxifen
- Tibilone doers not increase mammographic density
- Tibolone anti-tumor effect not direct anti-Estrogen (1)
- is not Er blocker
- does not inhibit aromatase in model systems
- placental aromatase on androstenedione (A -> E1) is not inhibited by tibolone
- effect of tibolone on breast aromatase is unknown
- see below for sulfatase effects, metabolite effects
- Tibolone anti-tumor effect not direct Androgenic (2)
- antiandrogen flutamide with tibolone does not reverse tumor inhibition (nude mice)
- [ISLT] no local formation of delta4 isomer in mice
- [ISLT] SBG increase mitigates androgenic effect
- Tibolone almost completely prevented initiation of breast tumor growth given at the time of induction***
- Tibolone anti-tumor effect is mostly Progestogenic (3)***
- cell proliferation is inhibited by tibolone
- apoptosis is stimulated by tibolone in normal breast cells and breast cancer cells
- delta4 isomer and pure progestin ORG2057 show similar apoptopic effects to tibolone
- Estrogenic metabolites of tibolone (3a-OH, 3b-OH)
- no mitogenic activities in normal breast epithelial cells
- even antiproliferative effects at high concentrations!
- stimulate apoptosis at high concentrations (different from estradiol)
- less expression of Ki67 and Pr (steroid secretion) than with estrogen seen in nude mice
- Tibolone and metabolites also inhibit sulfatase activity***
- 3-0H tibolone metabolites inhibit sulfatase in breast (E1S -> E1)
- may limit estrogenic effects of estrogen in breast
- sulfatase, sulfotransferase determine the amount of active estrogenic metabolites
- high levels of estrone sulfate are present in breast tissue (E1 -> E1S)
- source of active estrogens via sulfatase (E1S -> E1)
- major determinant of estrone sulfate to estradiol
- may also limit 3a-OH tibolone sulfate to active non-sulfate
- sulfated estrogens and 3OH-sulfated tibolone thus not hydolyzed to non-sulfated Er active forms in endometrium
- irreversible inhibition in some tumor lines by 3-OH metabolite
- already shown for tibolone in endometrium
- forms a low estrogenic environment, in which progestogens are known to inhibit breast proliferation
- [ISLT] low estrogen environment accentuates delta4 isomer effect
-
Sulfotransferase
activity may be induced by locally formed tibolone progestogenic forms in endometrium
- conversion to sulfated forms
- other progestins shown to induce sulfotransferase in endometrium
- Sulfotransferase (E1-> E1S), may also be induced by tibolone to limit active forms
- tested by Cherite
- non-genomic effect
- Androgenic activity
- delta4 isomer is androgenic
- [ISLT] possibility of androgenic effect of delta4 isomer in risk reduction
- delta4 isomer has direct androgenic effects
- androgens have an anti-estrogen effect in the breast
- MPA binds to androgen receptor in low estrogen environment
- [ISLT] MPA binds to androgen receptor in this situation
- [ISLT] Tibilone has both indirect anti-androgen and anti-estrogen activity
- decreases Testosterone and Estradiol
- 17BHSD-I weakly inhibited by 3-OH tibolone metabolites
- less E1->E2, less A ->Testosterone
- 17BHSD-II induced by tibolone
- more E2 -> E1, more Testosterone ->Androstenedione
- see fig 1 pg10 Steroid Interconversion Pathways
V. HORMONE REPLACEMENT THERAPY USE
IN RELATION TO TUMOR BIOLOGY AND
BREAST CANCER PROGNOSIS
[in Menopause - Hormones and Cancer Proceedings]
edited by M. Neves-e-Castro and B.G. Wren 2002
[MHC CH6 p49-53]
Authors
H. Olsson [MHC CH 6]
Department of Oncology
University Hospital
Lund S-221 85
Sweden
1999
- Hypothesis: age and differentiation of the breast epithelium at the time of initiation reflects tumor biology at eventual diagnosis
- more immature breast tissue gives more advanced tumors
- supported by findings that less advanced tumor stage has been found to be more common in HRT users
- more highly differentiated
- lower proliferation rate
- low histological grade
- p53-negative status
- more frequent estrgoen receptor positive status
- may be more lobular type tumor rather than ductal
- current HRT users may show a higher S-phase percentage
- Epidemiology
- highest risk for breast cancer seen for present use and low BMD
- [ANOTE] low BMD is associated with low serum estradiol
- Estrogen only seems to lower the risk
- Continuous Combined or Sequential Combined Estrogen effects from past studies are conflicting
- Sweden study shows increased breast cancer risk of diagnosis with
- breast cancer had 1.74 risk ratio with HRT
- no overall increased risk of total malignant tumors
- other cancers had lowered risk each alone was not statistically significant
- HRT was significantly associated with a lower risk of dying, longer survival (Odds Ratio = 0.78)
- notes
- Cox regression analysis similar to WHI
- newly incident tumors
- [ISLT] need mortality rates for that year
- [ISLT] need a combined "other tumor" group to compare for statistical significance
- Research indicates that tumor biology and short-term prognosis are more favorable in breast cancer patients who have used HRT prior to diagnosis
- better survival for current HRT users
- breast cancer survival
- total survival
- this is irrespective of tumor stage, hormone receptor status, and tumor detection mode
- no survival benefit seen after 10 years from stopping for past HRT users
- family history and HRT exposure appear to be independent risk factors for breast cancer incidence
- reduced sensitivity and specificity in breast cancer screening would be opposite to a screening bias
- screening bias- HRT group would screen more often and find more tumors
- sensitivity bias- HRT group would find fewer tumors when screened, due to increased density
- tumor size bias- HRT group would find more tumors when screened, due to tumors that are already there are bigger (estrogen is a growth hormone for breast tissue)
- these would influence incidence data
VI. HORMONAL THERAPY
FOLLOWING BREAST CANCER
[in Menopause - Hormones and Cancer Proceedings]
edited by M. Neves-e-Castro and B.G. Wren 2002
[MHC CH 7 p55-66]
Authors
B.G. Wren [MHC CH 7]
Sydney Menopause Centre
Royal Hospital for Women
Randwick
New South Wales
Australia
1999
- "The breast is capable of producing its own estrogen, independent of circulating blood levels of estradiol.
It is known that ,
breast [fat] cells are capable of converting steroid precursors into estradiol at levels many times greater than is found in circulating blood
and that
this local activity is probably the major source of hormones responsible for the promotion and growth of breast cancers
."
- "It is also acknowledged that
sex hormones are not responsible for initiating the ocogenic mutations that cause breast cancer
, but they do have a marked influence on the rate of mitotic activity echibited by normal and malignant cells."
- "The fact that women continue to seek a satisfactory preparation to relieve their estrogen deficiency-related distress is a clear indication that these alternative agents are not capable of controlling those menopausal problems."
- "The previously held belief that exogenous administration of hormones is contraindicated for post menopausal women with breast cancer may not always be so."
- Why Estrogen and Progesterone don't cause Breast Cancer
- Epidemiological results
- If estrogen were an initiating cause of breast cancer, would expect a declining incidence of tumors for Postmenopausal women who cease to produce Estrogen.
- The opposite is found.
- Women over the age of 70 years are ten times more likely to have breast cancer diagnosis than women under the age of 40.
- Beral and the Collaborative Group on Hormonal Factors in Breast Cancer have shown that the incidence increases in direct proportion to age, and in a reanalysis of 51 major studies showed a 1.023 per year of use increase.
- If estrogen or progesterone were the initiating cause of breast cancer, would expect an increased risk of recurrence or new tumors with the high levels of Estrogen and Progesterone in Pregnancy. No increased risk is seen, or even a reduction in recurrence risk or death.
- Studies SUPPORT HRT (decreased breast cancer risk)
- Some randomized studies show no increased recurrence or death when estrogen with continuous progestogen is given after diagnosis and treatment of breast cancer.
- NHANESI (Lando and coleagues) showed that women using HRT (mainly unopposed estrogen) for as long as 22 years had a RR = 0.9 (1-3 years), RR = 0.5 (3-9 years), RR = 0.9 (10 or more years). [cohort study]
- Schairer and co-workers reported a non- statistical 10% reduction in breast cancer risk with unopposed continuous progestins for 5 or more years. (see below for sequential estrogen-progestin) [cohort study]
- Ross and co-workers noted that continuous progestin given to postmenopausal women was not associated with an increased risk with a history of breast cancer. (see below for premenopausal) [retrospective cohort study]
- Plu-Bureau and associates found a 50% reduction in the risk of developing breast cancer when given unopposed progestogen for up to 10 years.
- Nachtigall and associates and Gambrell and associates showed a reduced risk of breast cancer with sequential progestogen.
- Ewertz in Denmark showed a non-significant reduction in breast cancer risk with use of progestogen.
- Studies OPPOSE HRT
(increased breast cancer risk)
- Persson and co-workers (1997) showed a 2-fold increase in breast cancer for women who were taking HRT. The risk was increased with the addition of progestin. This was not statistically significant however. [retrospective case-control]
- Colditz and colleagues and Goldstein and colleagues showed evidence from the epidemiological Nurses Health Study that suggested that estrogen increases the risk of breast cancer, with sequential progestin increasing the risk. [epidemiologic study]
- Ewertz in Denmark showed a slightly increased risk of breast cancer with the use of estrogen. (see above for progestin).
- Schairer and co-workers showed an increase of 20-30% on breast cancer with sequential estrogen-progesterone over 5 years or more. (see above for progestins) [cohort study]
- Ross and co-workers concluded a 10% increase in breast cancer risk for each 5 years of sequential estrogen-progestin use in premenopausal with a history of breast cancer. (see above for post menopausal). [retrospective cohort study]
- EXPLANATION for these findings***
- Bi-phasic response for Progestin (Musgrove and co-workers 1991)
- short term progestogen application increases breast cell mitosis
- continous progestogen application decreases breast cell mitosis through an inhibition of phosphorylation of retinoblastoma protein
- Mechanisms of Progestin on Breast Cancer***
- estradiol dehydrogenase and estrone sulfotransferase are induced by progestin, transforming active estradiol into inactive estrone sulfate
- matures alveolar breast cells which dont proliferate
- reduces estradiol receptors which inhibits cell mitosis
- reduces production of proto-oncogenes c-myc and c-fos, known to accelerate mallignant breast cell mitosis
- reduces production of cathepsin D, a potent breast cancer growth factor
- increases apoptosis and death of breast cells
- "Continous progestogen use (wth or without estrogen) has been shown to reduce the rate of breast cell mitosis, and this is likely to reduce the risk of breast cancer."
- "Druckman has shown that not only is there variation in the cellular response to different concentrations of progestogen, but some progestogens appear to have a potent inhibitory response while others have none."
- Estrogen effects on Breast Cancer***
- it is often forgotten that estrogen has been used to treat women with breast cancer with equal results to Tamoxifen
- women with pregnancy after breast cancer also have good or better long-term survival
- many processes for growth and death in breast cells affected
- mitosis
- angiogenesis
- cell differentiation
- quiescence
- repair
- apoptosis
- estrogens and progestins affect these cell signals
- cyclins
- cyclin-dependent kinases
- inhibitor proteins
- growth factors
- "The older concept, held by many physicians, that hormones are universally contraindicated for women with breast cancer, can now no longer be substantiated."
- "If oncogenic mutations occur following sporadic translocation of genetic material during mitosis, then it is reasonable to conclude that any increase in cell division will result in an increased rate of cancer."
- "Unopposed estrogen with or without sequential progestogens will increase the rate of breast cell mitosis; there is now clinical and epidemiological evidence that this increased mitotic rate is associated with an increased rate of cancer." Note however that this is an assumption.
- Study results from Royal Hospital for Women Study, Sydney Australia (Wren)
- There is no evidence for increased breast cancer recurrence when continuous Estrogen plus Progestogen is given to symptomatic Breast Cancer patients
in the Royal Hospital Study for over 25 years
(a population-based study starting in 1968)
- E+P decreases Breast Cancer Reccurence and Death [see table 2 p59]
- OR = 0.4 for risk of recurrence
- OR = 0.6 for risk of recurrence on HRT (0-5) years after diagnosis
- OR = 0.3 for risk of recurrence on HRT (>5) years on HRT after diagnosis
- OR = 0.1 for risk of death
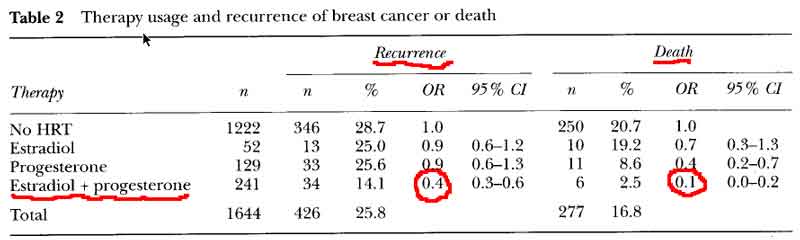
- no significant difference in new breast cancers for combined E+P HRT
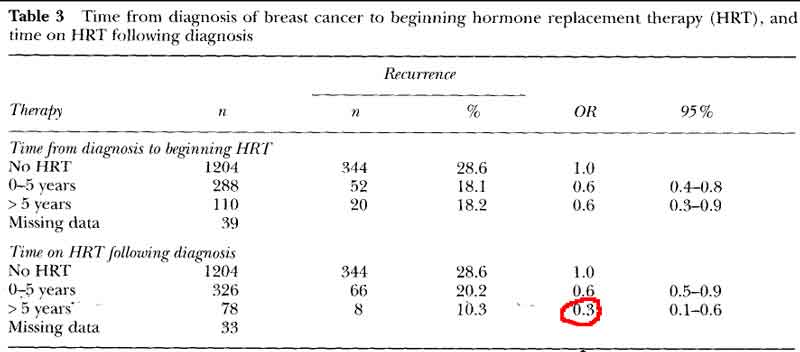
- women who began HRT within 5 years of treatment and continued therapy for many years had a significant reduction in the risk of recurrence or death.
- no advantage to delay beginning HRT till after 5years, with recurrence rates identical to the women who began HRT in the first 5 years.
- HRT decreases breast cancer recurrence even with delayed start
[see table 3 pg60]
- Comparison of Hormones after Breast Cancer
tamoxifen + estrogen,
tamoxifen + progestogen,
tamoxifen + estrogen + progestogen
all had lower rates of recurrence [see table 5 p61]
the best results were found when a progestogen and estrogen were added to tamoxifen, the same reduction seen for estrogen plus progestin alone (OR = 0.4)

- study characteristics
- retrospective (historical) cohort study
- after treatment for breast cancer, cohorts of 442 women elected to take HRT, 1222 women elected not to take
- continuous dosing
- most started within 5 years after treatment, 28% >5 years after treatment
- most were self-referred (symptomatic)
- majority of women who took unopposed hi dose progestogen did so for menopausal symptoms after developing metastases
- usually offered Provera 200-400mg or megesterol acetate 40-60 mg daily (high dose)
- initially offered a moderate dose of Provera 50mg or Northisterone acetate 5mg, but regimen has varied
- Provera dose ranging from 5 to100mg and norestisterone ranging from 1 to 5 mg
- 46 of 52 using unopposed estrogen were using a vaginal topical application, thought to have little effect on breast activity
- Premarin/conjugated estrogen 0.3mg or 0.625 mg, Ogen/estrone sulfate 1.25mg, estradiol valerate 2mg, or 17B-estradiol 1-2 mg was added for continued symptoms
- HRT users were average 4 years younger, with slightly smaller tumors, and more stage I, and more exposed to HRT prior to study
- Steps to inhibit Estrogenic activity have yielded the best results to date in terms of prolongation of life after Breast Cancer.
- All following regimens have resulted in a 30-40% improvement in survival time
- ovarian ablation
- anti-estrogens (tamoxifen)
- specific estrogen receptor modulators/ SERMS (raloxifene)
- estrogen antagonists
- aromatase inhibitors
- progestogens
- Hormones are often given given after breast cancer treatment with positive results
- Tamoxifen (weak estrogen) has been used for partial remission of breast cancer in up to 40% of women
- Progestins and SERMS reduce tumor growth
- Short term vs long term benefits?
- Tamoxifen >5 years increases risks
- progestins appear to have long term and possible short term benefits
- Summary about Continuous Hormone Dosing:***
"The clinical use of progestogen, particularly medroxy progesterone acetate (MPA), has been promoted for over 40 years to treat women with secondary spread of breast cancer. While the dosage of MPA has of necessity been high (400-2000mg) the inhibitory effect has been as effective as that of tamoxifen, and is probably mediated by the same internal cell-cycle control mechanisms which influence cell division in vitro. The theraputic applications of continuous progestin therapy has been shown to reduce the risk of new breast cancers and to inhibit the progress of breast cancer that has spread outside the breast.
However, in spite of the excellent effect that progestogens (MPA, megestrol acetate, norethisterone, dydrogesterone) have on reducing breast cell mitosis and inhibiting breast cancer growth, a large number of women using progestogen continue to remain distressed by estrogen deficiency symptoms and thus require the addition of estrogen to control their problems (Table 6). In the present study we found that about one-quarter of those women who had breast cancer elected to take estrogen with continuous progestogen to relieve symptoms of estrogen deficiency.
Following a review of this self-selected group of women, it would appear that those who chose to use this form of HRT were not disadvantaged by the therapy. There was no increase in either recurrence or deaths among women on HRT. In fact, women receiving combined estrogen-progestogen had a lower risk of recurrence or of death than women not on HRT, presenting a similar profile to those women who became pregnant following breast cancer. There was no advantage to women who delayed using HRT till after 5 years following their treatment for breast cancer, with recurrence rates identical to those of women beginning HRT in the first 5 years.
However, for women who began HRT within 5 years of treatment for breast cancer, and continued the therapy for many years after, there was a significant reduction in the risk of recurrence or death. It appears from these figures that, once therapy has been inititated and the longer it is maintained, the less is the risk of recurrence.
The risk of developing a new cancer while on continuous combined estrogen and progestogen was not increased above the risk found among women who did not use HRT, suggesting that continuous combined HRT is not responsible for initiating the development of breast cancer.
The main advantage for those women taking combined therapy lay in the fact that their distressing estrogen deficiency symptoms had been relieved without compromising their suvival outcome. This population-based comparative study suggests that estrogen and progestogen administered continuously to women with a history of breast cancer does not have an adverse outcome. From our results, it would appear that the use of continous progestogen in a hormonal regimen conveys a degree of protection from breast cancer development and growth that is not evident with unopposed estrogen or with sequential progestogen and estrogen."
[ISLT] Continuous Combined Hormonal regimen may be better for symptomatic patients (high estrogen, premenopausal status). It is possible that asymptomatic (postmenopausal) patients may do better with cyclic therapy (low estrogen status, low receptor status).
VII. PHYTOESTROGENS AND BREAST CANCER
[in Menopause - Hormones and Cancer Proceedings]
edited by M. Neves-e-Castro and B.G. Wren 2002
[MHC CH 8 p67-115]
Authors
H. Adlercreutz [MHC CH 8]
Department of Clinical Chemistry
PL 60, FIN-00014
University of Helisinki
Helisinki
Finland
1999
- The precursors of biologically active phytoestrogens detected in man are found mainly in soybean products, whole-grain cereal food, seeds, berries, tea, legumes and some vetgetables, such as carrots and Brussel sprouts.
- plant lignan and isoflavonoid glycosides found in these foods are converted by intestinal bacteria to hormone-like compounds.
- the weakly estrogenic diphenols formed have been shown to influence sex hormone production, metabolism, mechanism of action and biological activity, and influence many intracellular steroid metabolizing enzymes.
- compounds included in the group of phytoestrogens have changed over time because of new compounds detected which have weak estrogenic activity such as the mamalian lignands and some flavonoids and the elimination of the fungal or mycoestrogens, the resorcyclic acid lactones, from the group because they are not plant estrogens.
- other estrogenic plant compounds that do not have two phenolic groups and have completely different structures not included in this definition of phytoestrogens
- Isoflavones
- dietary isoflavones are found in soy. smaller amounts of isoflavones in other beans, peanuts, some vegetables, and fruits
- genistein (soy)
- daidzen (soy)
- glycitein (soy)
- coumestrol occurs in only a few foods not normally consumed in western culture, alfalfa sprouts and brussel sprouts
- the most estrogenic of these compounds are coumestrol, genistein, and equol (metabolite of daidzen, produced in small amounts by all, and large amounts by only about 30% of people both in western countries and in Asia)
- be aware of variation in laboratory assays
- enterodiol and enterolactone are formed from plant lignan glycoside precursors by the activity of gut microflora in the proximal colon. intestinal bacteria seem to play a significant role in the maintenance of the plasma enterolactone concentration.
- secoisolariciresinol is converted to enterodiol and this is converted to enterolactone (flaxseed)
- marairesinol is converted directly to enterolactone
- new enterolactone precursors are pinoresinol, laricirerinol, syringaresinol, 7-hydroxymatairesinol and arctigenin, the first three of which are indentified in rye bread
- urinary excretion of enterolactone
- increased with increased serum hormone binding globulin (SHBG)
- decreased with increased free estradiol and free testosterone
- increased with fiber intake
- Breast Cancer
- soy
- few effects found in prospective studies for soy in adults
- may be more significant neonatally and in children
- isoflavonoids in plasma and urine are good biomarkers for intake of soy products, but may not be the active components from the diet
- equol
- vegetarians and subjects who consumed more carbohydrates and less fat have higher equol excretion in urine
- likely that higher equol excretion reflected a more healthy diet
- high equol producers may have particularly low risk of breast cancer due to both reductions in sex hormone levels and alteration of endogenous estrogen metabolism
- [ISLT] progestogen-like effect
- positive effect may depend on estrogen levels
- reviews are mixed on effects of equol. may be more associated with isoflavones and lignans and the heathier diet which lowers risk, rather than directly related to breast cancer risk.
- low excretion of lignans was found in healthy breast cancer patients, along with the low equol excretion
- SHBG efects may be due to associated soy consumption (more free hormones though)
- associated grain consumption may increase enterolactone levels
- enterolactone plasma/ urine levels in individuals in a Finnish study
- intake of grain calories or grain fiber/kg body weight correlated with the excretion of enterolactone in urine
- correlation between grain fiber intake and urinary enterolactone was 0.707
- rye bread seemed to be the best source of lignans
- plasma levels were almost 3x higher after consumption of rye bread than with white bread
- both types of bread were consumed at least 200g/day with no other cereals permitted
- berries, fruits and vegetables also contibuted to lignan excretion
- smoking decreased plasma levels
- obstipation, whole grain food intake, vegetables, berries and fruits increased plasma levels
- a healthy lifestyle and diet seem to explain only a small part of the individual variations
- antibiotics immediately reduces the formation of mammalian lignans to very low levels for a long time, sometimes for more than a year.
- enterolactone levels were low in breast cancer individuals, higher in cancer free individuals
- countries with high legume consumption have lower disease rates***
- consumption of a lignan-rich food and fiber (which contains lignans) seems more consistently related to lowered risk than effect of dietary soybeans
- many different compounds other than soy (protease inhibitors, phytic acid, B-sisterol) are active compounds
- diet composition with regard to fatty acids, animal and plant protein, and various carbohydrates and fibers likely plays an essential role
- vegetarians have higher levels of isoflavonoids in plasma and urine
- in Hawaii, Japanese women excrete more isoflavones than Caucasian women
- subjects in low risk areas for breast cancer excrete high hevels of isoflavones and lignans
- chimpanzees in captivity are very resistant to breast cancer and excrete large amounts of isoflavonoids and lignans with high relative amounts of equol.
- increased risk
- breast cancer subjects excrete low amounts of isoflavones and lignans
- within 6 months of immigrating to Hawaii from the Orient , immigrants that still consuming a very low fat diet excreted roughly 1/10th of the amounts of isoflavonoids than Japanese women in Japan, similar to omnivore levels.
- adult animal studies
- soy
- no effects seen with soy alone
- no effect seen with soy and high isoflavonoids
- lower non-significant risk in groups receiving high isoflavonoid diet
- lowers SHBG in plasma (???progestogenic effect, other cofactors)
- flaxseed contains very high amounts of secoisolariciresinol (SDG)
- flaxseed oil and particularly the purified SDG seem to inhibit the growth of mammary tumors in experimental rat studies
- both initiation and promotion
- SDG converted to enterolactone and enterodiol
- neonatal/ prepubertal rat studies
- genistein
- given to rats prepubertally or neonatally (conception until 21 days postpartum) at physiologic or greater amounts gave protection against 7,12-dimethylbenz anthracene (DMBA)-induced experimental breast cancer
- concluded that genistein (Lamatiniere et al)
- enhanced breast cell differentiation
- stimulates transforming growth factor (TGFa)
- stimulates epidermal growth factor receptor (EGF)
- down regulates EGF-signaling pathway in terminal end buds and terminal ducts of adult mammary glands, which may resist breast tumorogenesis
- may have estrogenic effects, and high estrogen in pregnancy may increase or lower breast cancer and prostate cancer risk in offspring, but evidence not definitive
- phytoestrogens are readily transfered from mother to fetus
- phytoestrogens during pregnancy may have an antiestrogenic (?progestogenic) effect in the high estrogen environment
- phytoestrogen summary
- soy is at most slightly protective for breast cancer
- high soy doses increase invasiveness of breast cancer, soy metabolites are estrogenic
- soy may be beneficial early in life, protecting from breast carcinogens later in life
- a fiber and lignan diet seems protective for breast cancer
- enterolactone may be a good biomarker of decreased breast cancer risk, but the mechanism is unkown
VIII. PROGESTERONE THERAPY
AND BREAST CANCER
[in Menopause - Hormones and Cancer Proceedings]
edited by M. Neves-e-Castro and B.G. Wren 2002
[MHC CH 13 p117-121]
B. von Schoultz [MHC CH 13]
Department of Obstetrics and Gynecology
Karolinska Hospital
Stockholm S-171 76
Sweden
1999
Fine Needle Aspiration (FNA), mammographic density evaluation
studies of human breast tissue proliferation
- epidemiological data indicates a slight to moderate duration-dependent increase in the relative risk of diagnosing breast cancer during use of HRT and Hormonal Contraceptives. This normalizes after 5-10 years of discontinuation.
- Three major theraputic combinations
- estrogen only
- estrogen in cyclic combination with progestogen
- estrogen in continous combination with progestogen
- all have different effects in the endometrium and other target organs
- few studies comparing these for the breast
- the interpretation of epidemiological data is complicated by a lack of basic understanding as to how sex steroids influence the breast and regulate epithelial proliferation.
- in vitro
- estrogens enhance breast cell proliferation
- addition of progestogen may reduce this effect
- in vivo
- proliferation of breast epithelial cells is seen during the luteal phase when progesterone levels are high
- proliferation results from FNA
- there is an increased breast epithelial cell proliferation in women using combined oral contraceptives, with marked individual variation in response to treatment
- positive correlation of proliferation and circulating levels of levonorgestrel progestogen
- negative correlation of proliferation and levels of free testosterone
- mammographic density results
- reflects the relative amount of fat, connective and epithelial tissues
- high amounts of connective and epithelial cells increase density on mammogram
- Wolfe classification
- N1, normal breast, primarliy fat with at most ficrous connective tissue strands
- P1, prominent ductal pattern in up to 1/4 of the breast volume
- P2, prominent ductal pattern in more than 1/4 of the breat volume
- Dy, extrememely dense parenchyma which usually denotes connective tissue hyperplasia
- In this study, CEE+MPA(conjugated equine estrogens/medroxyprogesterone acetate) had
similar Wolfe increase from E2+NE(estradiol/norethinodrone)[see table1p118 all increase].

- Estrogen alone had little increase except at higher dosing [table2 p119 all increase]

- PEPI trial and others (cline/von schoulz) have reported that different regimens for HRT have different impacts on mammograhic density
- increase in density more frequent in women on combined estrogen-progestogen than on estrogen alone
- increase with estrogen alone is small (E)
- women with continuous combined show 40-50% with increased density (E/P continuous)
- many women have no increase in density, there is individual variation in response
- there is no consensus on the interpretation of breast density and histological findings in breast cancer
- may correlate with increased risk of tumor (stimulation)
- may make diagnosis of tumors more difficult (increased density hides tumors)
- increase in mammographic density appears to be an early event that occurs during the first few months of therapy, and thereafter remains stable during long term treatment with the same regimen
- [ISLT] would then be more likely to have suspicious mammogram requiring biopsy in previous users, and so would initially find more breast cancers than in group who did not have previous hormone exposure (surveillance/detection bias, comparison mammogram, do more biopsies, find more cancer that is already there)
- [ISLT] increase in density would hide tumors in the just initiating hormone group unless more likely to read as suspicious
- macaques monkey experimental model
- surgically postmenopausal macaques were given HRT
- proliferative response after combined treatment with conjugated equine estrogens plus medroxyprogesterone acetate was much more pronounced than for treatment with estrogen alone.
- progestogen augments response to estrogen thru several mechanisms depending on dosing/endogenous exposure
- would expect to see an initial proliferative response because hormone deficient
- [ISLT] would be similar to late menopausal status (loss of steroids from the ovary, particularly estrogen).
- there is no consensus about the proliferative effects of the addition of progestogen to estrogen treatment
- effects of progestogen vary by type, duration of exposure and the estrogenic environment
- both estrogen and progestogen in high doses have a growth supressing effect and have been used to treat breast cancer
- 5-10mg of norethindrone(northisterone) in combination with estrogen appears to reduce the rick of breast cancer recurrence
- norethisterone (norethindrone) is highly progestogenic and mildly androgenic
- [ISLT] progestin dose is too low in some studies and some HRT formulations
- [ISLT] hi Free Testosterone serum levels secondary to low SHBG decreases mamographic density and normal breast cell proliferation [see Peter Conner et al, Fertility and Sterility, 81(6):June2004]
IX. PROGESTINS AND BREAST CANCER RISK:
STATE OF THE CONTROVERSY
[in Menopause - Hormones and Cancer Proceedings]
edited by M. Neves-e-Castro and B.G. Wren 2002
[MHC CH 14 p123-128]
Authors
R. Sitruk-Ware and G. Plu-Bureau [CH 14]
Center for Biomedical Research
Population Control Council
1230 York Avenue
New York, NY 10021
USA
- The use of progestins with estrogen is a recent phenomena. Progestin has been used since the early 1980's in Scandinavia and the UK. The FDA recommended the addition of progestins to various estrogen regimens in 1990, and the use became widespread.
- non-differentiated terminal end buds are susceptible to malignant transformation by carcinogens
- carcinogen applied to immature undifferentiated terminal end buds, followed by administration of sex steroids, led to transformed cells and cancer
- sex steroids applied first to immature animals at the stage of undifferentiated terminal end buds led to maturation and differentiation of the alveolar buds, which did not undergo any malignant transformation when carcinogen was applied (ie, after the steroids had already cause differentiation)
- suggests that malignant transformation comes before sex steroid exposure, not after, and prior sex steroid exposure is protective against malignant transformation
- steroid effects
- Era receptors and Pr receptors are expressed in the same cells
- these cells act as steroid sensors and control proliferation through paracrine mechanisms onto other cells
- Estrogen stimulates growth of the breast ductal system
- very low Estrogen concentrations express Pr receptors
- estradiol (not progesterone) was the major mitogen in epithelial breast cells [Laidlaw and colleagues]
- high doses of estradiol are required to induce proliferation
- Estrogen also stimulates immature (Type 1) terminal bud growth, but not mature (Type 3 & 4) terminal bud growth
- long term estrogen therapy increased cell proliferation and the percentage of breast made up of glandular tissue
- Progestin acted as a proliferative agent with estrogen, transforming the ducts into differentiated (mature) lobules (branching)
- increase in the terminal duct lobular units is observed when progestin was used with estrogen
- progesterone transforms the terminal end buds into alveolar buds and differentiated lobules ready for milk secretion
- Type 1 immature lobules exhibit a high rate of cell proliferation (estrogen sensitive) [Russo and colleaques 2001]
- Type 2 lobules appear after pregnancy and evolve into highly differentiated Type 3 & Type 4 lobules with very low cell proliferation (estrogen insensitive)
- nulliparous women have up to 80% Type 1 lobules
- parous women have Type 3 lobules
- after menopause, breast tissue involutes
- under HRT, breast tissue grows again and Type 1 lobules reappear
- in parous women these lobules have markers of the previous differentiated (mature) state
- should not interpret tissue proliferation as necessarily indicating undifferentiated and transformed cells, rather than indicating normal cells
- combined growth effects of estrogen and progesterone
- low estrogen = "on your mark"
- progestin/estrogen = "get set, one cycle and wait"
- breast cells in the late phase of the cell cycle are driven into the S phase of DNA synthesis by progestins
- this effect is transient, with further application of progestin supressing the cyclins, halting breast cell division in the early G1 phase. [Musgrove and associates]
- high estrogen = "go"
- effect of estrogen, progestin on cancer risk summary (see above)
- estrogen only --> immature lobules/ ducts, more cancer risk
- estrogen/progestin --> mature lobules, low cancer risk
- balance between estrogenic and progestogenic components of HRT vary with the dose and regimen
- MPA is a derivative of progesterone
- 19-norprogesterone derivates (17OHprog ones) preferred for HRT in Southern Europe
- medroxyprogesterone actetate
- megestrol
- no direct estrogenic effects like NE
- exhibits only slight androgenic effects
- decreases SHBG
- less direct androgenic effects compared to 19-NorTestosterone derivatives
- [ISLT] MPA effect on Free Testosterone depends on functional status of ovary,
the net of two effects:
- increases Free Testosterone fraction from decreased SHBG
- decreases ovarian production of androgens more (treats hirsuitism)
- [ISLT] different effects depending on pre/peri/menopausal status
- exhibits glucocorticoid-like activity (displaces Free Cortisol from Cortisol Binding Globulin (CBG)
- (NE) norethindrone acetate (1x) and levonorgestrel (100x) have direct androgenic properties
- norethindrone also has weak estrogenic effect at Er receptors
- oral micronized estradiol or estradiol valerate lead to high circulating levels of estrone due to liver metabolism, with high total circulating levels of estrogen
- tumor proliferation
- results of proliferation testing is unclear, as breast cells would react to the cumulative effect of estrogens and progesterone secreted over several cycles, rather than to daily changes in hormone production
- different Progestins may induce different effects
- augment Er receptors vs reduce Er receptors
- the role of Progesterone could not be separated from the role of Estrogen
- MCF7 tumor cell proliferation (Catherino and co-workers)
- promegestrone (R5020) shown to decrease cell proliferation (in normal breast cells)
- MPA and nomegestrol acetate do not induce proliferation
- norgestrel and gestodene stimulate proliferation (Er receptor mediated)
- follicular phase study, women undergoing surgery for benign breast disease
- randomly treated with estradiol, progesterone or placebo, and mitoses in the epithelial cells in normal part of the breast was counted
- after estradiol tx, both estradiol concentration in the breast and number of mitoses was high (0.83/1000)
- in placebo, the number of mitoses was low (0.50/1000)
- after Progesterone Tx, Progesterone concentration was high, mitoses low (0.17/1000)
- authors concluded that in vivo, high intra-tissue concentrations of Progesterone decrease the mitotic activity of normal lobular epithelial cells
- [ISLT] implies a dose effect for progestin
- meta-review [Oxford 1997]
- reviewed most of the epidemiological studies
- 17,000 cancer cases; 32,000 controls
- breast cancer risk in HRT users was RR = 1.14 compared to non-users
- risk increased 2.3% per year of use
- current users at diagnosis, hormones for >5 years
- RR = 1.6 > 15 years
- increased risk for lean women (BMI >25 kg/m2) receiving HRT for more than 5 years
- no significant difference between the users of estrogen or combined estrogen plus progestin
- more recent studies
- in general
- indicated a higher risk from users of combined HRT
- percentage of combined HRT use was low (around 5%)
- 5 percent had unknown hormone therapy
- there was no information about the risk factors
- Ross and co-workers
- lower risk of breast tumors, used continuous HRT
- conjugated equine estrogen and MPA
- Magnussen and colleagues
- lower risk of breast tumors, used sequential HRT
- swedish cohort
- oral estradiol or estradiol valerate plus norethindrone or levonorgestrel progestin
- higher levels of total estogen (estrone up to 466 pg/ml) in those that received 2mg oral estradiol than other regimens
- premenopausal women with benign breast disease (Plu-Bureau)
- progestins appeared to be beneficial
- cohort study
- 1150 premenopausal French women with benign breast disease
- followup of 10 years
- progestin use and the duration of use were not found to be significantly associated with breast cancer risk
- linear decrease in breast cancer risk with duration of progestin use
- does not support the hypothesis that progestins increase the risk of breast cancer
- 19-nortestosterone derivatives were found to be significantly associated with a lower risk of breast cancer (RR = 0.48)
- [ISLT] use of 19-nortestosterone derivatives as off study medications in placebo group would lower breast cancer risk relative to Prempro group in WHI
- [ISLT] supports either continuous or adequate cyclic progestin lowers risk
- Trends in progestin consumption in the world
- USA lagged behind Europe in use of progestins in HRT
- 12x higher progestin use in France than USA in 1985, 3x in 1995
- lower risk of breast cancer in France trend seen compared to the USA
- curve analysis does not permit the establishment of any causal relationship between progestin consumption and breast cancer incidence however in population studies
X. HORMONES AND EPITHELIAL
OVARIAN CANCER
[in Menopause - Hormones and Cancer Proceedings]
edited by M. Neves-e-Castro and B.G. Wren 2002
[CH 15 p129-140]
Author
H.A. Risch [MHC CH 15]
Department of Epidemiology and Public Health
Yale University School of Medicine
60 College Street, P.O. Box 208034
New Haven, CT 06520-8034
USA
1999
- Androgens and Ovarian Cancer
- Plasma Estrone is produced by adipose aromatization of Androstenedione, about half of which is ovarian, and half adrenal in younger women
- Breast and ovarian epithelial cells express 17B-hydroxysteroid dehydrogenase (17BHSD), which converts Androstenedione to Testosterone (and Estrone to Estradiol)
- Androstenedione is a Weak Androgen, which does not bind strongly to Ar Androgen receptor
- converted in epithelial cells to Testosterone, which does bind to Ar
- Plasma levels of Androgens are greater than those of Estrogens, even during the late follicular phase of the menstural cycle, when Estrogen levels are highest.
- Androgens may stimulate epithelial cell proliferation
- Testosterone stimulates ovarian epithelium
- epidemiology supports the association between Androgens and risk of Ovarian Cancer
XI. NEUROSTEROIDS and SYNTHESIS
[in Menopause - Hormones and Cancer Proceedings]
edited by M. Neves-e-Castro and B.G. Wren 2002
[MHC CH 1, p1-7]
Authors
E. Plassart-Schiess and E.E. Baulieu [MHC CH 1]
INSERM U488 and College de France
Hopital du Kremlin-Bicetre
94276 Le Kremlin-Bicetre
France
1999
Location, Synthesis and Function of Neuro/Sex steroids
(Underneath the Hood)
- Neurosteroids are found in nervous system as well as reproductive organs, and provide additional information that may be relevant to the understanding of reproductive steroid function and cancer
- [see fig1 p2] Steroid Synthesis
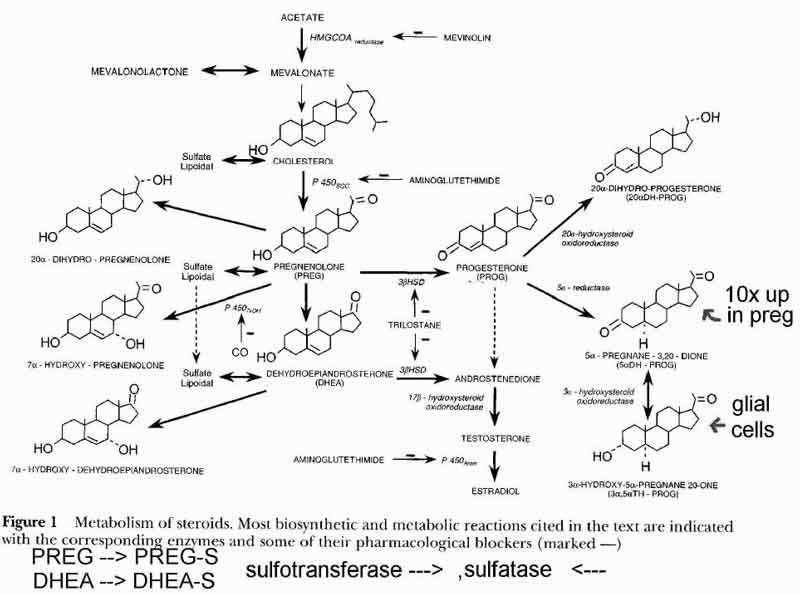
- in the mitochondria compartment (1)
- acetate is converted to cholesterol
- thru HMGCOAreductase
- cholesterol is starting substance for neuro/reproductive/adrenal steroids
- acetate is obtained from sugar sources (3 carbon from 6 carbon)
- outer membrane (receptors)
- GABA receptors
- benzodiazepines, ETOH
- chloride channel, allows Cl entry into neuron
- hyperpolarizes and dampens neuron excitability (depressant)
- delta subunit inhibits steroid modulation
- allows cholesterol into inner membane
- PregenoloneS and DHEAS inhibit GABA effects (excite neurons)
- TH-Prog enhances GABA effects (depresses neurons)
- ETOH ???
- NMDA receptors
- pregnenoloneS potentiates NMDA receptors
- may reinforce pregnenonloneS antagonism of GABA
- inner membrane
- synthesis of precursor steroids
- cholesterol converted to precursor steroids pregnenolone(nPREG), DHEA
- thru sccP450 in oligodendrocytes and schwann (glial) cells
- neuropregnenolone (nPREG) function
- microtubules
- memory
- myelin repair
- in the endoplasmic reticulum compartment (2)
- conversion of nPREG and DHEA to intermediate steroids (P and A, delta5) and (Androstenedione, delta4)
- 3B-HSD
- regulated by cell density, high cell density inhibits
- low activity in glial and neurons
- Sigma-1 receptors
- DHEAS is a sigma-1 agonist
- PregenonloneS is a sigma-1 inverse agonist (antagonist???) p 4 [ ]
- Progesterone is a sigma-1 antagonist
- in the cytosol compartment (3)
- further metabolism of intermediate steroids
- conversion to sex steroids (E and T)
- conversion to active sulfate and fatty acid esters
- conversion to TH-P (tetrahydroprogesterone) in glial cells thru DH-P (dihydroprog)
- microtubules
- important for growth and maintenance of neurites during neuronal differentiation
- composed of tubulin and MAPs
- MAPS (microtubule-associated proteins)
- Maps major components of neuronal cytoplasm
- regulate microtubule lattice formation and dynamics
- determine neronal shape and control the balance between regidity and plasticity in neural processes
- MAP1, MAP2, TAU families
- Pregnenolone (MAP2) receptors
- Map2 receptors (microtubule-associated protein type2)
- MAP2A, MAP2B (hi molecular weight)
- MAP2C, MAP2D (lo molecular weight)
- limited to dendrites
- Pregnenolone acts at level of microtubules via MAP2 receptors
- co-polymerization with tubulin favors pregnenolone binding
- accelerates microtubule polymerization, especially at low MAP2 concentrations
- increases mictrotubule amounts, are normal appearing
- Taxol causes abnormal appearing microtubules
- PregnenoloneS and Progesterone have similar affinity to Pregnenolone
- competitive inhibitors at Pregnenolone receptor (inactive on MAP2)
- so cell growth effects mediated thru MAP2/mictotubules
- [see fig 3a p 6]
- Pregnenolone (stimulates)
- Pregnenolone-S (inhibits)
- Progesterone (inhibits)
- DHEA, DHEAS,Testosterone, Cortisol, Estradiol
- different mechanism on microtubules than pregnenolone
- have low affinity at MAP2 receptors (not MAP2)
- DHEAS only negligible, DHEA weak competitors at Pregnenolone receptor
- DHEAS increases dendrite length containing MAP2 marker
- DHEA increases dendrite length containing TAU marker
- so these ovarian and adrenal hormones have no MAP2 effects on neurons (except Progesterone, Preg, PregS
- 2-methoxyestradiol is a major estradiol metabolite***
- 2-methoxyestradiol inhibits cell growth
- hi 2-methoxyestradiol = inhibits microtubule polymerization at high concentrations [MHC p 5]
- low 2-methoxyestradiol = allows microtubule polymerization, but abnormal morphology similar to Taxol effect at low concentrations
- binds directly to purified tubulin without MAP (polymerized and unpolymerized by glutamate)
- doesnt act at pregnenolone receptor (MAP2), so different mode of action from pregnenolone
- ???1/2 life
- in the nucleus compartment (nuclear steroid receptors) (4)
- E receptors/ Estrogen
- E2 induces P receptors
- bind circulating and hypothalamic (local in brain) E
- stimulates growth and differentiation
- P receptors/ Progesterone
- E2 induces Pr receptors in hypothalamic neurons, oligodendrocytes (not cortex)
- P decreases Pr receptors
- P production stimulated by adjacent neurons
- P receptors can also activate without ligand by phosphorylation
- P inhibits E growth and differentiation in oligodendrocytes and astrocytes, thru binding to P receptors
- nuclear receptor binding
- progesterone synthesis
- P stimulates myelin protein synthesis in schwann glial cells
- sciatic nerve Schwann cells
- dont know if nerve and peripheral receptors the same [p3]
- Neurosteroid Hormone Overview
- major steroid metabolite forms affecting breast cancer
- free steroid (active)
- sulfate esters (inactive)
- converted to sulfate esters by sulfatase
- free hormone from sulfate esters by sulfotransfersase
- sulfate metabolites modulate GABA, NMDA, Sigma1 receptors
- autocrine/paracrine (local)
- fatty acid esters (inactive)
- specific hormones
- Pregnenolone (PREG)
- most abundant and active neurosteroid in rat brain (with PregS)
- acts at level of microtubules
- via pregnenolone receptor (MAP2 microtubule-associated protein type2)
- [see fig2 p3]
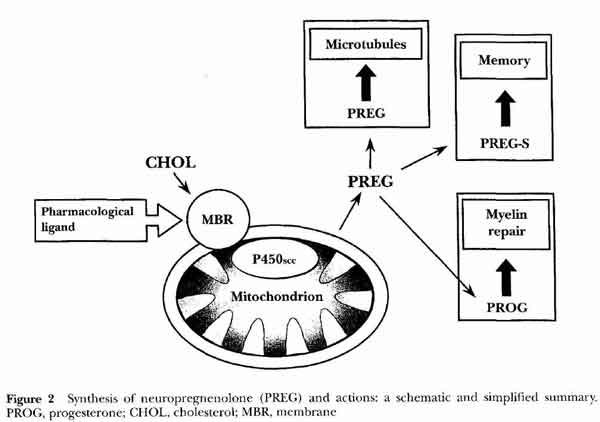
- Pregnenolone Sulfate (PREG-S)
- most abundant neurosteroids in rat brain (with Preg), most active
- binds to pregnenolone receptor
- enhances GABA
- potentiates NMDA receptor
- sigma-1 inverse agonist
- Progesterone (PROG active)
- sigma-1 antagonist
- progestins
- medroxyprogesterone acetate
- norethindrone actetate
- TetrahydroProgesterone (TH-P)
- by 3a-oxioreductase, in glial cells (helper actions, stimulate myelin???)
- enhances GABA
- DHEA (active)
- DHEAS (inactive)
- stimulates GABA effect
XII. WHERE ARE WE NOW?
[in Menopause - Hormones and Cancer Proceedings]
edited by M. Neves-e-Castro and B.G. Wren 2002
[CH 16 p141-145]
Author
M. Neves-e-Castro [CH 16]
Reproductive Medicine Clinic
Av. Antonio Augusto de Aguiar
24-2o.DT
Lisbon 1050
Portugal
1999
- Large retrospective and prospective epidemiological studies have reported that:
- With HRT for 10 years there are only six more cases of breast cancer diagnosed per 1000 women. This increased annual risk is equivalent to one year of delayed menopause after the age of 50 that is observed in women who have never used HRT.
- For the same 10 year treatment with HRT, it is likely that six cardiovascular events may be averted.
- HERS study (Heart and Estrogen/progestin Replacement Study) concluded it was not advisable to adopt HRT for the secondary prevention of cardiovascular events. The protective role of estrogens had no solid support.
- The opposite was seen in the Nurses Health Study (Grodstein and colleagues), which indicate that Estrogens have a clear benefit for primary prevention of heart disease.
- "Unfortunately, many of these studies are biased, misinterpreted and misquoted." The patient populations are also different.
- "The benefits [of menopausal hormones] are clear in the short and long term, not necessarily for every woman, but certainly for most of them... Estrogens are probably the only available molecules having nerve growth activity at the central nervous system level in addition to enhancing the activity of many neurotransmitters.
Also, they offer protection against colon cancer, they stimulate synthesis of nitrous oxide in the vascular endothelium, and may act as calcium channel blockers,
they prevent oxidation of low-density lipoprotein cholesterol and stimulate the production of high density lipoprotein, and they stimulate osteoblasts and decrease the activity of osteoclasts.
Finally, they are not harmful if properly monitored for potential, though rare, side effects."
back
www.DrTimDelivers.com
[an error occurred while processing this directive]page views since March2008
*This page is combined from original posted pages and reformatted on 3/09/2008, with the
Graphic of Tumor Estrogen and its author note added 3/09/2008.